


Urea cycle disorders are rare, genetic conditions that affect the body’s ability to remove ammonia from blood. When the urea cycle isn’t working properly, ammonia builds up in the blood and can cause vomiting and confusion. This is why it’s so important to have a doctor diagnose you right away.
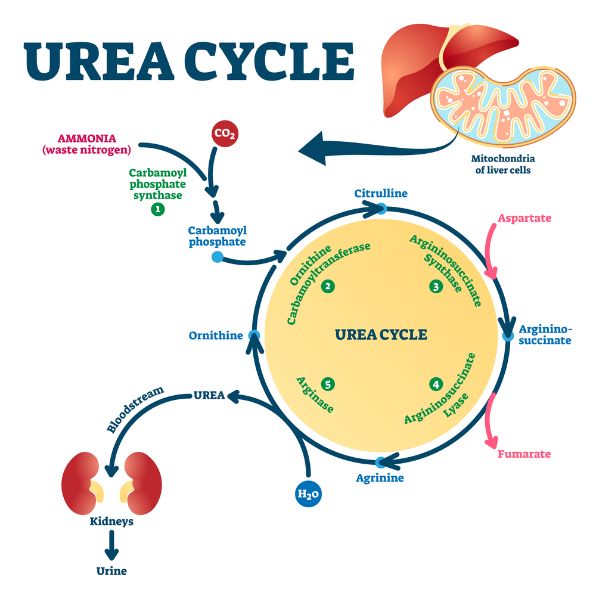
UCDs are caused by a problem with one of the six enzymes or two transporters of the urea cycle pathway (CPS1, OTC, ASS1, ASL, NAGS, and citrin). The severity of a urea cycle disorder depends on which gene is affected.
Severe urea cycle disorders occur in newborns, usually within 36 to 48 hours of birth, and can lead to death. These infants develop anorexia, alterations in respiratory function, hyperventilation, hypothermia, seizures, and neurologic posturing.
Mild urea cycle disorders occur later in life, usually after a stressor such as infection, surgery, or dietary protein load interferes with the urea cycle. They may have subtle symptoms that go undiagnosed or unheeded and can lead to permanent brain damage.
The majority of UCDs occur in children and are caused by a defect in any of the enzymes or transporters of the urea cycle pathway. These enzymes are able to break down proteins, and they help the body eliminate ammonia from the blood.
Individuals with severe urea cycle disorders have no enzyme activity in the pathway and ammonia builds up rapidly. These symptoms can include deteriorating neurological status, seizures, and coma.
Some urea cycle disorders are hereditary, which means that the condition is passed down from parent to child through an autosomal recessive pattern. The parents of a child with these disorders have one normal gene and one abnormal gene, which causes the defect.
These genes can be tested by a blood test. The result of the test is an estimate of the number of copies of the abnormal gene that are in the body. The more abnormal genes there are, the greater the risk of having a child with a urea cycle disorder.
If a person has an inherited urea cycle disorder, their children have a 25 percent chance of also having the same condition. The parents of a child who has an inherited urea cycle disorder will need to be tested to determine whether they carry the defective gene.
There are a variety of treatment options for urea cycle disorders, including diet, medication, and liver transplantation. Most patients with urea cycle disorders will need to have their ammonia levels monitored by doctors throughout their lives.
The diagnosis of a urea cycle disorder can be made by blood tests and laboratory testing. In some cases, it can be confirmed by a biopsy of the intestine or the liver.
Treatment of urea cycle disorders involves lowering ammonia concentrations by reducing dietary protein and by using alternative pathways to remove ammonia. The goal is to keep the ammonia in the blood low enough that it doesn’t cause vomiting or other problems.
Treatment can be complicated, as there are multiple factors that affect a patient’s ammonia levels. Many of these patients will need to be treated by specialists in metabolic disease. These doctors will give a patient strict dietary instructions, supplement the body with l-arginine and l-carnitine, and possibly use scavenging agents.


{kind=link}